Machine learning methods that predict protein fitness from sequence remain sensitive to changes in data distributions,
limiting generalization across common conditions encountered in protein engineering. Practically, protein engineers
are thus left wondering about the effective utility of ML tools. The FLIP benchmark established protocols for testing
generalization under some domain shifts, but it was limited to measurements of thermostability, binding, and viral capsid viability.
We introduce FLIP2, a protein fitness benchmark spanning seven new datasets, including enzymes, protein-protein interactions,
and light-sensitive proteins, as well as splits that measure generalization relevant to real-world protein engineering campaigns.
Evaluating a suite of benchmark models across these datasets and splits reveals that simpler models often matched or outperformed
fine-tuned protein language models on FLIP2, challenging the utility of existing transfer learning techniques. Provenance for all
datasets has been recorded and we redistribute all data under CC-BY 4.0 to facilitate continued progress.
7 new protein datasets spanning enzymes, protein-protein interactions, and light-sensitive proteins
16 challenging splits that test generalization across mutations, positions, fitness levels, and wild-types
Real-world engineering scenarios including position-based, mutation-based, and wild-type-based splits
Comprehensive baseline evaluations with zero-shot pLMs, ridge regression, and fine-tuned models
Open access with CC-BY 4.0 licensing and complete provenance tracking
Overview
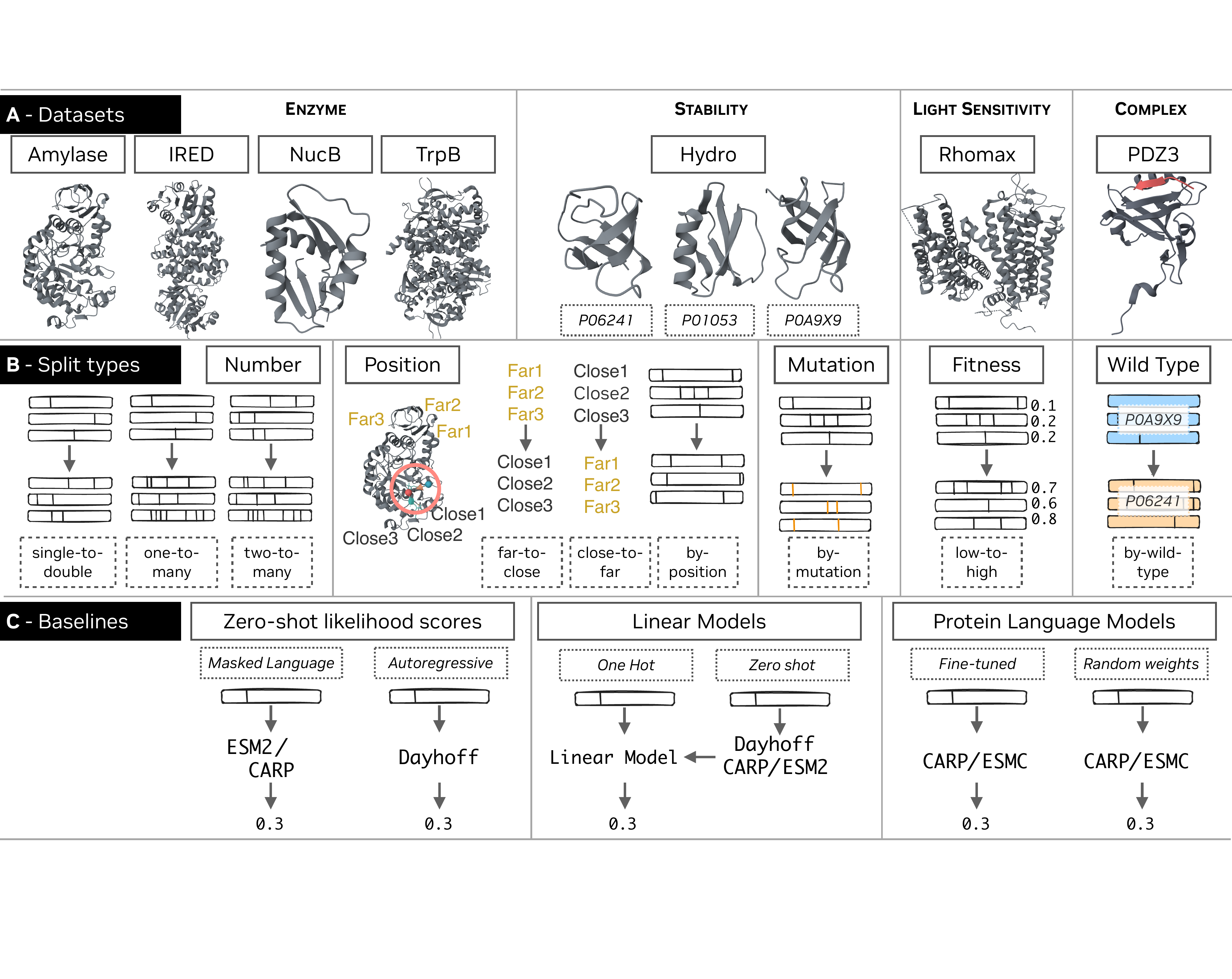
Figure 1: The FLIP2 benchmark. (A) The FLIP2 datasets. Solid boxes indicate dataset names, dashed boxes indicate individual protein identifiers.
(B) The FLIP2 splits (dashed boxes) and split types (solid boxes). (C) Baseline sequence-to-fitness prediction methods.
FLIP2 significantly expands the original FLIP benchmark with new datasets that better represent the diversity of protein
engineering applications. While FLIP focused on thermostability, binding, and viral capsid viability, FLIP2 introduces
datasets covering enzymatic activity, protein-protein interactions, and optogenetics applications.
The benchmark includes 16 distinct train-test splits across 5 categories that simulate realistic protein
engineering scenarios: generalization to more mutations (number splits), to mutations at different sequence positions
(position splits), to unseen mutations (mutation splits), to higher fitness variants (fitness splits), and to different
wild-type proteins (wild-type splits).
The FLIP2 Datasets
FLIP2 includes seven datasets covering diverse protein functions and engineering challenges. Each dataset contains
experimentally measured fitness values for hundreds to hundreds of thousands of protein variants.
Alpha Amylase (Amylase)
Alpha amylases catalyze the breakdown of starches and are used in detergents to remove starch stains. This dataset
studies stain removal activity for variants of Bacillus subtilis alpha amylase, including variants with
up to eight mutations.
3,706Total Variants
488Single Mutants
4Splits
Splits:
one-to-many (number): Train on variants with 1 mutation; test on all others
close-to-far (position): Train on mutations within 7.3Å of active site; test on mutations farther away
far-to-close (position): Train on mutations far from active site; test on mutations closer to active site
by-mutation (mutation): Train and test on different mutations
Source: Van der Flier et al., Computational and Structural Biotechnology Journal (2024) License: MIT
Imine Reductase (IRED)
Imine reductases reduce imines to amines and are employed in pharmaceutical production. This dataset contains
activity measurements from a microfluidic screen of Streptosporangium roseum imine reductase variants
from an error-prone PCR library, including variants with up to 15 mutations.
17,143Total Variants
1,207Single Mutants
1Split
Splits:
two-to-many (number): Train on variants with 0, 1, or 2 mutations; test on all others
Source: Gantz et al., Microdroplet screening study (2024) License: CC-BY 4.0
Nuclease B (NucB)
Endonucleases such as Bacillus licheniformis Nuclease B (NucB) degrade DNA and have potential applications
in chronic wound care by degrading extracellular DNA required for biofilm formation. Wild-type NucB activity drops
to around 80% at physiological pH. This dataset measures nuclease activity at pH 7 for variants from an error-prone
PCR library. To ameliorate assay noise effects, measurements are binned into four activity levels.
55,760Total Variants
4Activity Bins
1Split
Splits:
two-to-many (number): Train on variants with 0, 1, or 2 mutations; test on all others
Source: Thomas et al., Engineering nuclease design (2025) License: CC-BY 4.0
Tryptophan Synthase β-Subunit (TrpB)
The β-subunit of tryptophan synthase (TrpB) synthesizes tryptophan from indole and serine and is essential for
cell growth. This dataset contains growth-based fitness measurements for combinatorially complete landscapes across
multiple sets of interacting residues, comprising ten different sub-landscapes across 20 positions.
228,298Total Variants
10Sub-landscapes
3Splits
Splits:
one-to-many (number): Train on variants with 0 or 1 mutations; test on all others
two-to-many (number): Train on variants with 0, 1, or 2 mutations; test on all others
by-position (position): Train on 3-site landscapes with no positional overlap; test on 4-site landscape and remaining 3-site landscapes
The hydrophobic core of a protein is crucial for its function and stability. This dataset contains stability measurements
for variants of three proteins (UniProt entries P06241, P01053, P0A9X9) where seven core residues in each protein
were randomized to hydrophobic amino acids (phenylalanine, isoleucine, leucine, methionine, and valine).
24,935Total Variants
3Wild-type Proteins
5Splits
Splits:
three-to-many (number): Train on variants with 0, 1, 2, or 3 mutations; test on all others
low-to-high (fitness): Train on variants below median fitness; test on variants above median
to-P06241 (wild-type): Train on P01053 and P0A9X9 variants; test on P06241 variants
to-P0A9X9 (wild-type): Train on P01053 and P06241 variants; test on P0A9X9 variants
to-P01053 (wild-type): Train on P0A9X9 and P06241 variants; test on P01053 variants
Rhodopsins are light-activated membrane proteins with applications in optogenetics. This dataset contains peak
absorption wavelength measurements for variants and chimeras derived from 75 microbial rhodopsin sequences.
Note that 41 wild types have no variants, while the remainder have between 1 and 181 variants, including
sequences with between 1 and 6 mutations and chimeras.
884Total Variants
75Wild-types
1Split
Splits:
by-wild-type (wild-type): Train on 5 most common wild-types; validate on 34 wild-types; test on 36 wild-types. Each split has a similar mean absorption wavelength.
Source: Karasuyama et al., Inoue et al., Sela et al. (2018-2024) License: CC-BY 4.0
PDZ Domain (PDZ3)
PDZ domains can bind to short linear motifs in intrinsically disordered regions (IDRs). This dataset measures binding
affinity between mutant PDZ3 domains and mutant CRIPT peptides. From over 200,000 assayed double mutation pairs,
the test set is filtered to 579 sequence pairs exhibiting significant non-additive binding effects (epistasis),
where observed affinity significantly exceeds predictions from a simple additive model.
734Variant Pairs
200,000+Total Assayed
1Split
Splits:
single-to-double (number): Train on single PDZ domain mutations; test on double mutations (one in PDZ3 and one in CRIPT peptide) exhibiting epistasis
FLIP2 includes five categories of splits that test different types of generalization relevant to protein engineering:
Number Splits
Train on variants with fewer mutations and test on variants with more mutations. Tests ability to extrapolate
to larger numbers of mutations where data becomes sparse.
Train and test on variants with mutations in different positions in the sequence. Tests ability to generalize
to previously unperturbed positions targeted in subsequent engineering rounds.
Examples: close-to-far, far-to-close, by-position
Note: Position splits are among the most challenging in FLIP2.
Mutation Splits
Train and test on variants with different unique mutations. Mutations to different amino acids at the same
position may be split across train and test sets.
Examples: by-mutation
Fitness Splits
Train on variants with lower fitness and test on variants with higher fitness. Simulates optimization campaigns
where later variants have higher fitness.
Examples: low-to-high
Note: Fitness splits are among the most challenging in FLIP2.
Wild-type Splits
Train and test on variants with different wild-type sequences. Tests ability to transfer mutational effects
across homologous proteins, critical when limited data exists per wild-type.
Note: Wild-type splits are among the most challenging in FLIP2.
Split Difficulty
Evaluation on FLIP2 shows that wild-type, position, and fitness splits are much more challenging
than number and mutation splits. This indicates that while models can generalize well to more mutations or
different mutations at the same position, they struggle to generalize to new positions or new protein backbones.
Baseline Performance
We evaluated zero-shot protein language model likelihood scores, ridge regression baselines, and fine-tuned pLMs
on all FLIP2 splits. Performance is measured using Spearman rank correlation and normalized discounted cumulative
gain (NDCG) on held-out test sets. These baselines confirmed that FLIP2 splits are more challenging than random
splits with the same number of training examples.
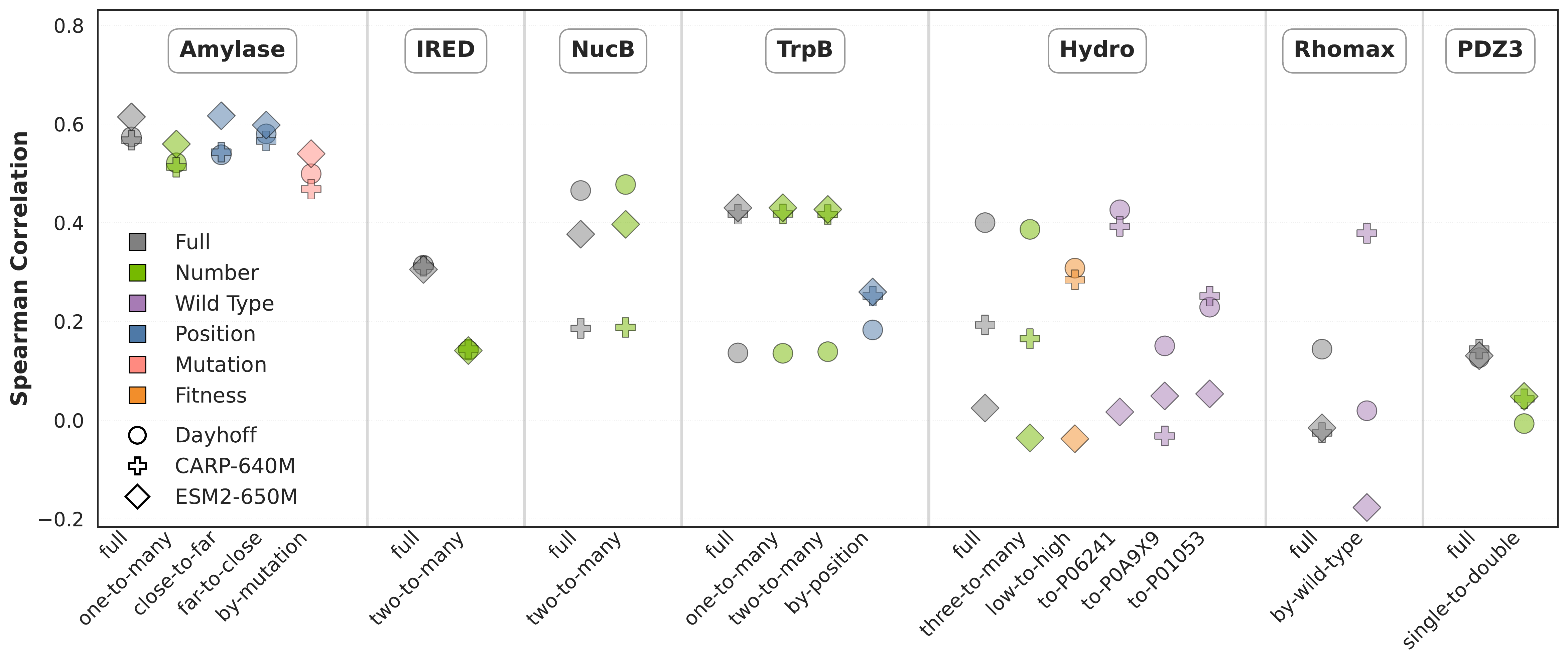
Figure 2: Zero-shot pLM likelihood scores on FLIP2 datasets and splits. Performance varies widely
across split types, with wild-type and position splits particularly challenging. Zero-shot likelihood scores are
more predictive for single-wildtype datasets (Amylase, IRED, NucB, TrpB) than for multi-wildtype datasets
(Hydro, Rhomax) or protein-protein interaction datasets (PDZ3). No single model performs best across all tasks.
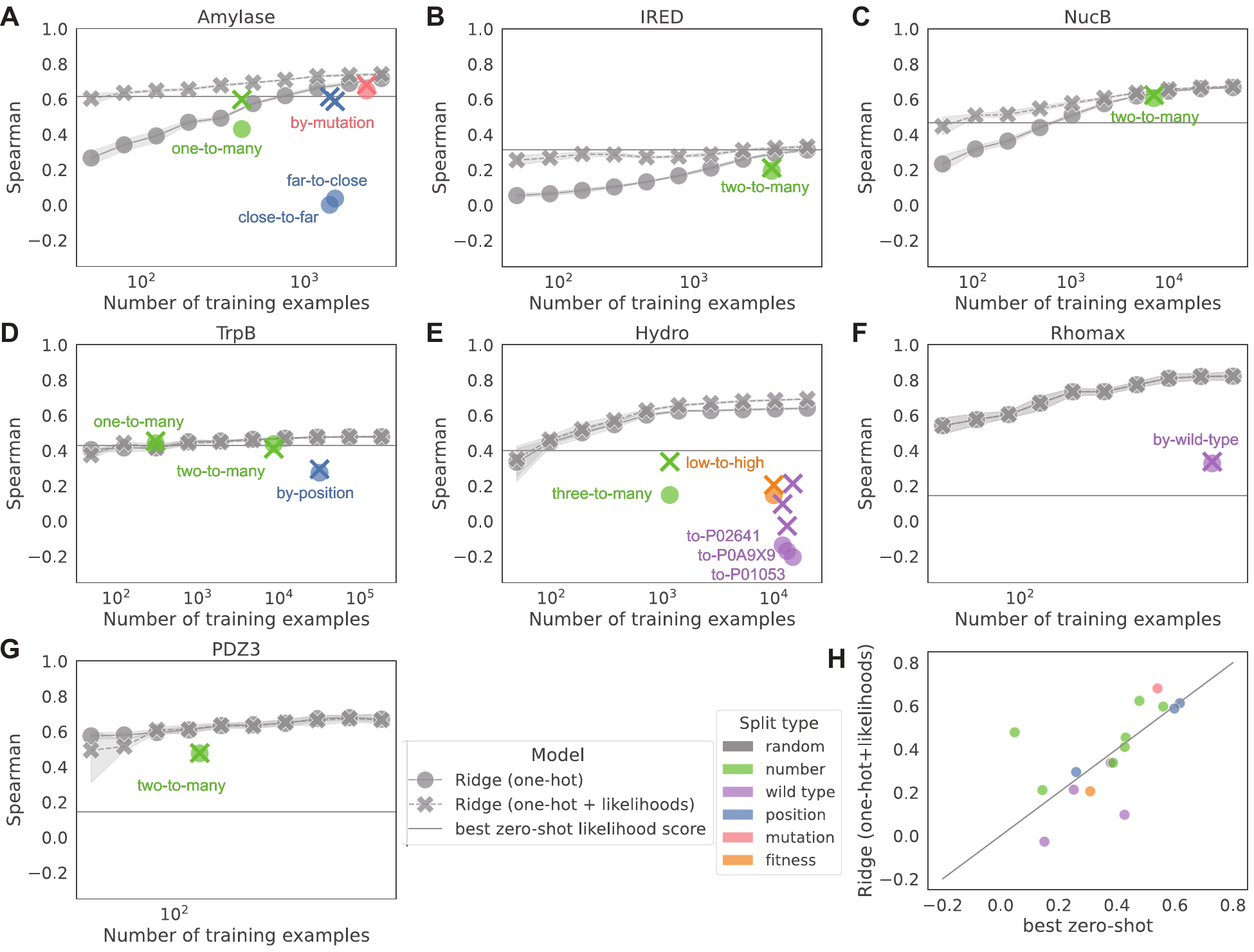
Figure 3: Ridge regression performance on each FLIP2 dataset and split. Spearman rank correlation
between ridge regression predictions and fitness for random splits of each landscape (gray points) versus FLIP2
splits (colored points). Ridge regressions were performed with only sequences as inputs (one-hot) or with sequences
and pLM likelihood scores as inputs (one-hot + likelihoods). Horizontal lines indicate the performance of the best
zero-shot pLM likelihood score for that landscape. FLIP2 splits are more challenging than random splits, with
wild-type, position, and fitness splits being particularly difficult.
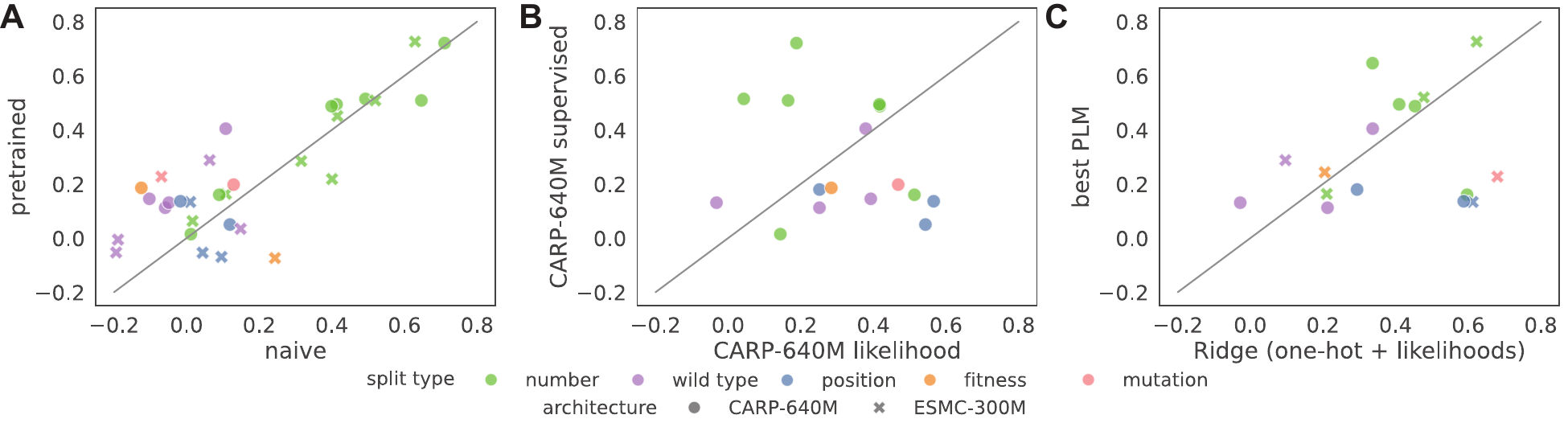
Figure 4: Fine-tuned pLM performance on each FLIP2 split. (A) Spearman rank correlation when
fine-tuning pLMs with and without pretraining. (B) Comparison of zero-shot CARP-640M likelihood scores versus
supervised fine-tuning. (C) Comparison of ridge regression with sequences and pLM likelihood scores versus the
best fine-tuned pLM for each split. Fine-tuning provides modest improvements over zero-shot scores, but simpler
ridge regression models often achieve competitive performance.
Key Findings
FLIP2 splits are more challenging than random splits with equivalent training set sizes, with wild-type, position, and fitness splits being substantially more difficult than number and mutation splits
Zero-shot pLM likelihood scores are effective for single-wildtype datasets (Amylase, IRED, NucB, TrpB) but struggle with multi-wildtype datasets (Hydro, Rhomax) and protein-protein interaction datasets (PDZ3)
Simple ridge regression on one-hot sequence representations often matches or outperforms fine-tuned pLM performance, especially when augmented with zero-shot likelihood scores
No single pLM architecture dominates across all datasets and splits - pLM choice is task dependent
Wild-type and position-based splits pose the greatest challenge for current methods, with fine-tuned pLMs showing limited improvement over simpler baselines
Fine-tuning with pretrained weights improved performance on only 14/16 tasks for CARP-640M and 9/16 tasks for ESMC-300M, challenging the utility of existing transfer learning techniques
Data Formats
FLIP2 datasets are provided in two formats: CSV for regression/classification tasks and FASTA for sequence-based tasks.
All data files are gzip-compressed (.csv.gz and .fasta.gz) for efficient storage and transfer.
CSV Format (Gzipped)
CSV files (*.csv.gz) contain four columns:
sequence: Amino acid sequence of the variant
target: Measured fitness value (continuous or binned)
set: Data split assignment ("train", "test", or "validation")
validation: Boolean indicating if the sequence is in the validation set
Example Python code to load FLIP2 data (gzipped files):
import pandas as pd
# Load gzipped CSV format (pandas handles .gz automatically)
df = pd.read_csv('assets/splits/amylase/one_to_many.csv.gz')
# Split into train/test
train_df = df[df['set'] == 'train']
test_df = df[df['set'] == 'test']
validation_df = df[df['validation'] == True]
# Alternative: Load FASTA format
from Bio import SeqIO
import gzip
with gzip.open('assets/splits/bind/one_vs_many.fasta.gz', 'rt') as f:
for record in SeqIO.parse(f, 'fasta'):
# Parse metadata from header
metadata = dict(item.split('=') for item in record.description.split()[1:])
sequence = str(record.seq)
target = float(metadata['TARGET'])
split = metadata['SET']
Download Data
All FLIP2 datasets and splits are available for download. All data files are gzip-compressed for efficient transfer.
Each dataset includes a README with complete provenance and attribution information. All data is licensed under
CC-BY 4.0 or MIT (see individual README files).
The original FLIP benchmark (2021) included datasets for AAV capsid viability, GB1 domain stability/binding,
and thermostability across multiple protein families. These datasets remain available for continuity and comparison.
Note: These legacy datasets are not part of the FLIP2 benchmark but are included for
researchers who wish to compare with the original FLIP results or use them for other purposes.
Citation
If you use FLIP2 in your research, please cite:
@article{didi2026flip2,
title={FLIP2: Expanding Protein Fitness Landscape Benchmarks for Real-World Machine Learning Applications},
author={Didi, Kieran and Alamdari, Sarah and Lu, Alex X. and Wittmann, Bruce and Johnston, Kadina E. and Amini, Ava A. and Madani, Ali and Czeneszew, Maya and Dallago, Christian and Yang, Kevin K.},
journal={Forty-third International Conference on Machine Learning},
year={2026}
}
Please also cite the original FLIP paper:
@article{dallago2021flip,
title={FLIP: Benchmark tasks in fitness landscape inference for proteins},
author={Dallago, Christian and Mou, Jody and Johnston, Kadina E and Wittmann, Bruce J and Bhattacharya, Nicholas and Goldman, Samuel and Madani, Ali and Yang, Kevin K},
journal={Thirty-fifth Conference on Neural Information Processing Systems Datasets and Benchmarks Track},
year={2021}
}
Individual Dataset Citations: Each dataset has specific attribution requirements. Please see the
README file in each dataset directory for proper citations and licenses.